


Mitochondria and the Electron Transport Chain (Part 3)
The fundamentals of mitochondrial genetics and clinical manifestations of mitochondrial disease


Image 1 - this image comes from the Child Neurology Foundation (https://www.childneurologyfoundation.org) and shows the many organ systems that can be adversely affected by mitochondrial problems.
Mitochondrial disorders often result from defects in respiratory chain complexes, as well as defects in various pathways that require mitochondrial function such as replication, translation, transcription, fusion, ribosome assembly, and activity.
The prevalence of mitochondrial disorders is estimated to be about 1 in 5000 across all ages. The clinical manifestations are often complex and can involve all body systems with high energy organs such as the brain, eyes, liver, muscles, and nerves being at particular risk.
The hallmark of any mitochondrial disease is the variability of the disease process within the same family or individuals despite similar mutations, which can complicate the diagnosis.
Clinical Manifestations and Common Symptoms
Some mitochondrial disorders can affect a single organ such as the eye seen in the mitochondria disease called Leber Hereditary Optic Neuropathy (LHON).
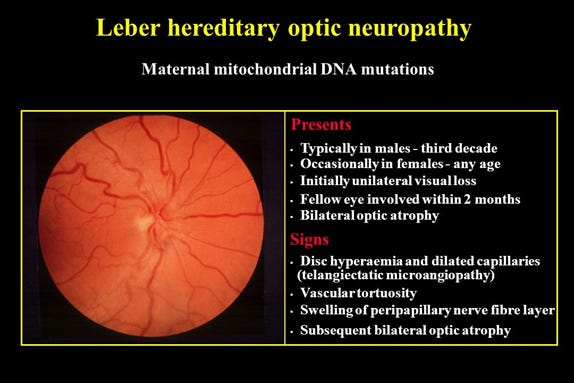
Image 2 - this image is from Slide Player - https://slideplayer.com/slide/8734957/
Most mitochondrial disorders involve multiple organ systems and present with both neurologic problems such as seizure, and myopathic issues which include muscle weakness.
Mitochondrial disorders can occur at any age with many mitochondrial DNA (mtDNA) conditions presenting in childhood, and nuclear DNA (nDNA) mitochondrial problems in manifesting in adulthood.
Some of the more common symptoms seen with mitochondrial disorders include:
Cardiomyopathy
Deafness
Diabetes mellitus
Exercise intolerance
External ophthalmoplegia
Optic atrophy
Pigmentary retinopathy
Ptosis
With regards to neurological manifestations, the following list of symptoms is often seen:
Ataxia
Chorea
Encephalopathy
Dementia
Migraines
Stoke-like episodes
Mid-to-late pregnancy loss is a common problem seen in mitochondrial disorders
Mode of Inheritance

Image 3 - mitochondrial inheritance occurs by maternal transfer of variants. Diseases caused by pathogenic variants in mitochondrial DNA can appear in every generation for a family and in both men and women. Fathers do not pass these disorders to their daughters or son.
Mitochondrial disorders may be caused by mutation of a mitochondrial DNA (mtDNA) gene or mutation of a nuclear gene (nDNA). The mtDNA pathogenic variants are transmitted by maternal inheritance (aka mitochondrial inheritance), whereas as nDNA pathogenic variants may be inherited in an autosomal recessive, autosomal dominant, or X-linked manner. Certain mitochondrial disorders involve deletion defects (loss of gene sequencing) which can be from an autosomal dominant or autosomal recessive manner.
The following image is of Autosomal Recessive Inheritance Pattern.

Image 4 - autosomal recessive inheritance can lead to deletion defects in gene sequencing. Often deletion defects are very serious in their manifestations. Notice in autosomal recessive inheritance that 25% of offspring are affected.
Mitochondrial Genetics

Image 5 - The mitochondrial genome contains 22 transfer RNA-encoding genes and 13 protein-encoding regions. The 13 encoding regions are highly specific for critical aspects of electron transport chain.
The mitochondria contain their own genome with each mitochondrion having DNA (mtDNA) in a circular double-helix containing about 16 kilobases (16 thousand base pairs – 16,569 to be exact). The mtDNA contain 13 protein-encoding regions involved in protein subunits of the respiratory complexes: I, III, IV, and V.
Notice that complex II associated with succinic acid dehydrogenase is not encoded by mtDNA. This enzyme, which is linked to the Krebs cycle too, is encoded by nDNA. Most proteins necessary for mitochondrial function are encoded by genes in the cell nucleus and the corresponding proteins are imported into the mitochondrion.
Mitochondrial DNA (mtDNA) versus Nuclear DNA (nDNA)
There are some differences in mtDNA and nDNA in what proteins that code for, and which electron transport chain complex (ETC) they are linked to:
mtDNA
DN1-6 regions code for complex I
CYT b codes for complex III
CO I and CO III codes for complex IV
ATPase 6 and 8 code for complex V
There are two ribosomal RNA genes and 22 transfer RNA genes that provide for intra-mitochondria protein synthesis. The advantage for a mitochondrion to produce some of its own proteins is so it can react independently of other cellular mitochondria, and to not solely be reliant on proteins produced by nuclear DNA. This allows for on-demand protein synthesis within a mitochondria, particularly in association with important subunits of the ETC.
nDNA
The bulk of the ETC proteins are produced from encoding genes within the nucleus of the cell. These proteins are critically important for overall ETC function and maintenance. Notice Coenzyme Q₁₀ is synthesized by nDNA which means it must be transported from the cytosol of the cell into the mitochondria and incorporated into complex I, II, and III. The following is a short list of nDNA influences on mitochondria:
Mitochondrial DNA maintenance
Mitochondrial protein synthesis
Coenzyme Q₁₀ synthesis
Respiratory chain complexes
Respiratory chain assembly factors
Mitochondrial structure
Conclusion
Mitochondrial diseases are a complex array of different phenotypic presentations involving multiple systems, many of which are linked to high demand organs: brain, ears, eyes, gastrointestinal, liver, kidneys, musculoskeletal, and nerves.
These diseases all involve problems within the respiratory chain, aka electron transport chain, and can be linked to not only mitochondrial genetics (maternal inheritance), but often influences from nuclear genetics as well. Mitochondrial diseases can manifest at any age, but most often their presentation of symptoms occurs during infancy or adolescence.
Not everyone with a mitochondrial problem has a mitochondrial disorder, aka disease. In fact, most people seen in a general practice, including functional and integrative medicine, are dealing with secondary mitochondrial problems, aka dysfunction, brought on by exogenous exposures such as environmental chemicals and mycotoxins or endogenous imbalances linked to a nutrient deficiency or metabolic problem.
You are not likely going to cure an individual of a primary mitochondrial disease, but it is possibly to improve their overall function and well-being. This is my goal in writing this Substack series of articles is to build a collective knowledge base to help more individuals dealing with a spectrum of mitochondrial problems.