


Mitochondria and the Electron Transport Chain (Part 4)
Recap of electron transport chain activity and complex I deficiency

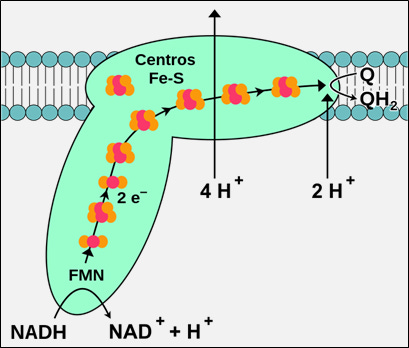
Image 1 - complex 1 of the electron transport chain. It has a characteristic L-shaped structure.
In the preceding three articles of this series on the mitochondria and the electron transport chain (ETC), we have gone over some fundamental aspects of the mitochondria, e.g. structure, and its function. In this article, I will provide more details about complex I, the largest complex of the ETC, and when severely deficient accounts for approximately 30% of all mitochondrial disorders.
To get things started, here is a brief overview of the electron transport chain again.

Image 2 - the 5 protein complexes of the electron transport chain. Notice that complexes I and II have a characteristic L-shaped pattern.
The ETC, found within the inner mitochondrial membrane (IMM), contains:
4 large primary protein complexes: I, II, III, IV.
2 independent components: ubiquinone, cytochrome c.
1 ATP synthase (aka complex V).
Iron-sulfur clusters (complexs I, II, III).
Electrons (e⁻) are conducted through the ETC in a specific sequence from reduced coenzymes such as NADH to oxygen.
The free-energy change drives the transport of protons (H⁺) from the matrix into the intermembrane space (IMS) via three proton pumps (complex I, III, and IV).
Electrons are funneled into the ETC by flavoproteins.
The ETC contains 4 flavoproteins:
Complex I – FMN (flavin mononucleotide).
Complex II, III, IV – FAD (flavin adenine dinucleotide).
These flavoproteins all reduce ubiquinone (Q, Q₁₀, or CoQ₁₀) at the beginning of the complexes:
Flavoprotein (reduced) + Q Flavoprotein (oxidized) + QH₂ (ubiquinol form of CoQ₁₀).
Protons (H⁺) are pumped into the IMS by complexes I, III, IV with oxygen (O₂) being the final electron acceptor which then gets reduced into water (H20).
Complex I (nicotinamide adenine dinucleotide (NADH):ubiquinone oxidoreductase)
The complex I enzyme is the first and largest enzyme of the mitochondrial respiratory chain (RC), and the oxidative phosphorylation (OXPHOS) system. It plays critical roles in transferring electrons from NADH (reduced) to coenzyme Q₁₀ (CoQ₁₀, ubiquinone), and in pumping protons (H⁺) to maintain the electrochemical gradient across the IMM.
Complex I is the largest complex of the ETC with 46 protein subunits. It’s basic function, includes:
Removes 2 electrons (e⁻) from NADH.
Transfers the e⁻ to ubiquinone (oxidized CoQ₁₀, Q).
A two step process reduces Q to ubiquinol (QH₂).
Pumps protons (H⁺) into the IMS.
Because of its size, complex I is the primary site of electron leakage and generation of superoxide free radicals, a type of reactive oxygen species (ROS). Some ROS are beneficial for mitochondrial signaling and function, but too much ROS causes damage for complex I and other ETC protein complexes leading to reduced adenosine triphosphate (ATP) production.
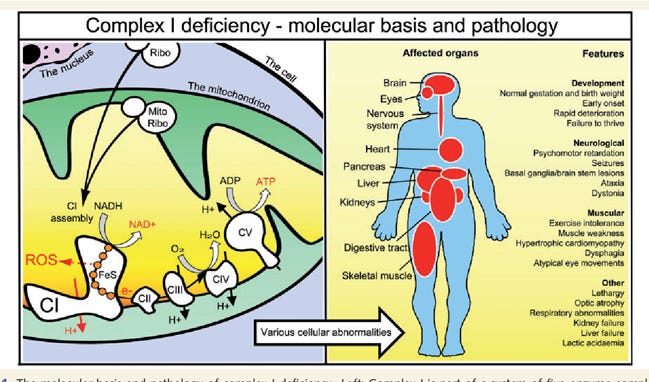
Image 3 - affected organs and features of isolated complex I deficiency - https://www.semanticscholar.org/paper/Assembly-factors-as-a-new-class-of-disease-genes-I-Nouws-Nijtmans/28a0eb05a0000a16e37b4b6ef9c229a5963ed789.
Isolated Complex I Deficiency
An isolated complex I deficiency is the most common identified defect in childhood-onset mitochondrial disease, accounting for approximately 30% of all cases of OXPHOS disorders.
Many children develop dramatic symptoms (see image 3 above) during the first year of life, and can have rapidly progressive disease. Unfortunately, for many of these children the disease of isolated complex I deficiency is fatal. However, clinical presentations vary which can range from fatal neonatal lactic acidosis to infantile-onset Leigh syndrome, childhood-onset mitochondrial encephalomyopathy, and other conditions.
The genetic defects of isolated complex I deficiency are complex and can result from both mitochondrial DNA (mtDNA) mutations and nuclear-encoded subunit and assembly factor mutations. Genetic defects have been reported for all 7 mtDNA-encoded complex I subunits, 17 of the 38 nuclear-encoded subunits, and multiple assembly factors.
For a review of mitochondrial genetics and clinical manifestations of mitochondrial diseases, see article 3 in this series.

Image 4 - reactive oxygen species causing electron transport chain complex disruption, and triggering other cellular reactions such as apoptosis - https://www.researchgate.net/figure/Link-between-oxidative-stress-mitochondrial-calcium-handling-and-cell-death-C-I-C-IV_fig4_359722454.
Vulnerability of Complex I
Not everyone with a complex I deficiency has a specific mitochondrial disease. In some cases, environmental exposures can lead to problems. Rotenone (a natural pesticide) is a complex I inhibitor, and likely other environmental compounds of similar chemical structure, may impeded its function.
High levels of ROS (which can occur from environmental chemicals and mycotoxins) can damage the complex too, along with other ETC proteins. In considering the vulnerability of complex I to ROS damage and subsequent deficiency, there are a few key things to remember:
Complex I pumps 4 protons into the IMS which accounts for approximately 40% of the electrochemical gradient to drive ATP production.
The L-shaped structure of complexx I contains 3 module sections: N, P, Q
N & Q subunits, which are nuclear-encoded, are in the peripheral arm of the protein complex protruding into the matrix. The P module is mostly within the inner mitochondrial membrane.
The N module is the input section for electrons and the Q module is for electron output.
Electrons from NADH are passed to flavin mononucleotide (FMN) to iron-sulphur clusters (Fe-S) to CoQ₁₀. This last step occurs near the junction of the peripheral arm with the membrane arm (P module):
Complex I contains 8 Fe-S clusters, the most of any complex within the ETC.
The conformational change that occurs between the peripheral and membrane arm with electron transfer allows for 4 protons to pump from the matrix into the intermembrane space.
There are 31 nuclear-encoded subunits for complex I that provide structural and regulatory activity. These subunits also act a “shield” around the core subunits (critical for its function) for electron transport and against oxidative stress.
The matrix is an area of the mitochondria with high oxygen tension where molecular oxygen is waiting to be converted to water through the actions of complex IV. It’s not difficult to recognize that environmental compounds that could damage sensitive regions within the nuclear DNA could alter the function of complex I. The same is true for mitochondrial DNA that provide specific proteins necessary for complex I function. As the protein complex becomes deficient it can no longer keep up with electron transfer, and the electrons leak into the matrix generating reactive oxygen species.
Conclusion
Complex I is critical for overall mitochondrial function and the generation of ATP through ETC activity. Because of its unique structure, size, and propensity to leak electrons it’s susceptible to damage and malfunction. A further complexity of complex I is its tendency for form supercomplexes, aka respirasomes, with complex III or even IV. Complex III is the second greatest area in the ETC for electron leakage. These supercomplexes do serve a purpose in combining actions as a whole for a mitochondrion to reduce its vulnerable for loss of function and it prevent sole activity independent of other complexes.
From a therapeutic standpoint there are many things to consider for complex I support. Most of the conventional literature references riboflavin and CoQ10. Therapies that help in mitochondrial biogenesis and antioxidant support can be helpful:
Vitamin B2 - 50mg/day (neonates), and 100mg to 300mg/day (adults).
CoQ10.
PQQ (pyrroloquinolone quinone) - check out my Substack article on PQQ and mitochondria.
MitoKatlyst - (-)-epicatechin.
Various antioxidants such as Sulforaphane and Ergothioneine - check out my Substack article on Longevity Vitamins.
Ketogenic diet.